Glaucoma is a leading cause of blindness worldwide and although current treatments usually stem the progression of the disease, there are many cases in which this approach is unsuccessful. In two animal models of glaucoma, we elevated intraocular pressure by either episcleral vein cauterization (in rats) or angle closure (in mice) and observed a dramatic elevation of the cytokine TNF-α, activation of microglia1-2, and a delayed, progressive loss of RGCs, thus mimicking key features of the disease. In one study, we showed that deletion of the gene for TNF-α or one of its receptors, or use of an antibody to TNF-α greatly slowed or arrested the loss of RGCs despite persistently elevated intraocular pressure1. In the second study, we showed that Etanercept, an FDA-approved soluble decoy receptor for TNF-α, arrested microglial activation at the optic nerve head along and stemmed the loss of RGCs2. These studies have contributed to a growing awareness of the role of TNF-α and inflammation as potentially critical factors in the pathophysiology of glaucoma.
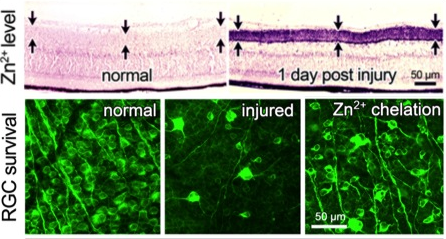
In other work, although optic nerve injury only directly affects RGCs, we found that it in fact causes profound changes throughout the retina. Within an hour after the optic nerve injury, we can observe a rise in ionic zinc (Zn2+) in the synaptic terminals that amacrine cells make onto RGC dendrites. Zn2+ is then exocytosed to alter RGC physiology3. Zn2+ accumulation in amacrine cell terminals requires the Zn2+ transporter ZnT3. Upstream changes events leading to this accumulation include reversal of glutamate transport in bipolar cells, NMDA receptor-mediated activation of the enzyme nitric oxide (NO) synthase-1 (NOS1) in a subset of amacrine cells, and elevation of NO. NO appears to react with reactive oxygen species to form peroxynitrite, which presumably reacts with metallothioneins to liberate Zn2+ 4-5. Chelating Zn2+ enhances the survival of RGCs and promotes a modest level of axon regeneration3, thus pointing to Zn2+ accumulation as one of the factors that contributes to RGC death and regenerative failure after optic nerve injury. On the other hand, preliminary studies suggest that NO may also play a positive role in enabling pro-regenerative treatments to stimulate regeneration (studies with Dr. Paul Rosenberg, Dr. Yiqing Li, Hui-Ya Gilbert, and other lab members).
References
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. J Neurosci Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma 2006;26(49):12633-41. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17151265
- Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, Benowitz LI, Miller JW. PLoS One Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma 2012;7(7):e40065. http://www.ncbi.nlm.nih.gov/pubmed/22802951.3388998
- Li Y, Andereggen L, Yuki K, Omura K, Yin Y, Gilbert HY, Erdogan B, Asdourian MS, Shrock C, de Lima S, Apfel UP, Zhuo Y, Hershfinkel M, Lippard SJ, Rosenberg PA, Benowitz L. Proc Natl Acad Sci U S A Mobile zinc increases rapidly in the retina after optic nerve injury and regulates ganglion cell survival and optic nerve regeneration 2017;114(2):E209-E218. https://www.ncbi.nlm.nih.gov/pubmed/28049831.PMC5240690
- Y. Li, et al. Presynaptic zinc regulates optic nerve regeneration: role of ZnT-3 and nNOS. Society for Neuroscience 2015. Chicago, IL, US.
- Y. Li, et al. Retinal Zn2+ elevation after optic nerve injury suppresses retinal ganglion cell survival and axon regeneration: upstream mechanisms involve GLT-1 mediated glutamate efflux, NMDA receptor activation, and NO generation. Society for Neuroscience 2016. San Diego, CA, US