Carroll Laboratory Research | Overview
The lab’s research interests are focused in two broad areas, i.e. autoimmunity and neuro-immunology.
Autoimmunity
SLE is an incurable autoimmune disease characterized by anti-nuclear antibodies (ANA). One current view is that disease is initiated by a “rogue” B cell that escapes negative selection and secretes pathogenic antibody against nuclear antigen eventually leading to formation of germinal centers and expansion of autoimmunity to include antibodies to many tissue antigens such as skin and kidney. Disease is amplified by activation of the complement system and secretion of inflammatory cytokines. In order to understand how self-reactive B cells are regulated, we use murine models of autoimmunity and conditional reporter mice that bear deficiencies in genes involved in normal adaptive immunity.
Some important questions in the field are:
- What initiates the escape of tolerance by a Rogue B cell?
- How does the dysregulation of a Rogue B cell result in chronic autoimmunity?
- What are the events that lead to spreading of the initial response to other tissue antigens?
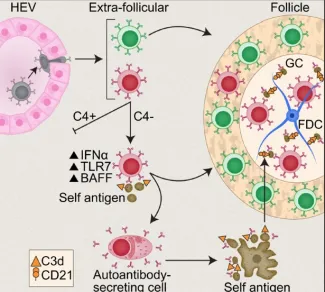
Figure 1. Immature self-reactive B cells that escape the bone marrow and enter the spleen are excluded from the follicles (“follicular exclusion”) and are eliminated. Under certain conditions such as excess type I interferon, triggering of TLR7 or absence of complement C4, a “Rogue B cell” can mature, express pathogenic autoantibody and enter the follicles where it can develop and chronically express pathogenic autoantibody. Red B cell-autoreactive; Green B cell- naïve normal B cell.
Neuroimmunology
It is increasingly evident that the brain is not isolated from events in the peripheral immune system and that innate pathways important in host protection can become dysregulated leading to neuropsychiatric changes. For example, increased copy number of the human complement C4A gene is a major risk factor for Schizophrenia; while in the periphery, it is protective against systemic autoimmunity such as lupus. Elevation in peripheral levels of interferon alpha (IFN-a) such as in viral infection, autoimmunity, interferonopathies or treatment with recombinant cytokine are strongly associated with psychiatric symptoms. We propose that blood-brain barrier entry of inflammatory cytokines such as IFN-a can activate multiple cell types in the brain triggering excess pruning of synapses altering neural circuits critical in executive function and cognitive memory.
Some important questions in the field are:
- Does “too much” expression of C4A promote loss of synapses leading to schizophrenia?
- What are the cell sources and regulation of the complement system in the brain?
- Does entry of IFN-alead to targeting of specific neural circuits by activated microglia?
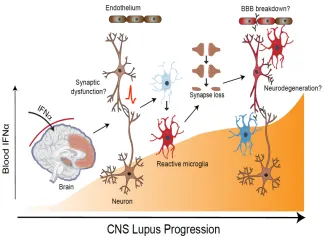
Figure 2. Inappropriate entry of interferon alpha into the brain can activate multiple cell types including neurons and microglia resulting in excess synaptic pruning and synapse loss. Pathology can be exacerbated by breakdown of blood brain barrier and leakage of additional serum proteins including antibody.
Tools used in the lab
Transgenic and conditional reporter mice, long term intravital 2-photon imaging of lymphoid tissues and CNS using implanted window chambers, scRNA seq for whole GC and memory B and T cell whole transcriptomes and BCR and TCR, multiplex in situ hybridization, flow cytometry analysis and cell sorting, parabiosis, construction of mouse models of disease using Crispr-cas 9 gene editing and BAC transgenic mice.